
Background and overview[1]
(1R,3S,4S)-3-(6-bromo-1H-benzimidazol-2-yl)-2-azabicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester can Harvoni is used as an intermediate in pharmaceutical synthesis, such as the preparation of a two-combination combination of the new antiviral NS5A inhibitor Ledipasvir (GS5885) and the NS5B blocker Sofosbuvir. It is a blockbuster hepatitis C treatment drug approved by Gilead in December 2013. Harvoni is the first all-oral anti-HCV regimen approved for the treatment of genotype 1 HCV infection that does not require concomitant interferon or ribavirin. Harvoni can be used alone or in combination with other oral preparations such as ribavirin.
Preparation[1]
(1R,3S,4S)-3-(6-bromo-1H-benzimidazol-2-yl)-2-azabicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester was used Synthesized by the following steps:
Step S1: First obtain compound 2 according to the following synthesis route;

Add 4.0g of 2-bromofluorene, disperse it in 175mL of glacial acetic acid, add 16mL of 20% sulfuric acid, 0.7g of potassium iodate, and 2.24g of elemental iodine, and heat and react at 80°C for 24 hours. Turn off the heating, cool the reaction in an ice bath, add 165 mL of cold water to dilute the reaction mixture, filter with suction, and wash the filter cake with tap water until the filtrate is colorless. Collect the obtained solid, vacuum dry it at 60°C overnight, add 40 mL of 3:1 (volume ratio) methylene chloride and methanol mixture, stir and beat under heating in an oil bath at 60°C for 1 hour, cool naturally overnight, filter, and vacuum the solid at 60°C. After drying overnight, compound 2 was obtained as a white solid (4.37g).
1HNMR(75MHz,DMSO)δ7.98(s,1H),7.87(s,1H),7.80(s,1H),7.75(d,J=0.8Hz,2H),7.58(dd,J= 8.2,1.8Hz,1H),3.95(s,2H).
Step S2: First obtain compound 4 according to the following synthesis route;
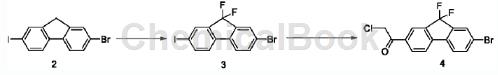
Take 21.96g of the compound, add 6.66g of N-fluorobisulfonamide to 40mL of anhydrous tetrahydrofuran under nitrogen protection, dissolve it, cool in an ice-salt bath for 30 minutes, and then add bistrimethyl dropwise within 20 minutes. Potassium silamide (1M tetrahydrofuran solution) was added dropwise and the temperature was raised naturally for 3 hours. Cool the reaction solution in an ice bath, add 0.1 mL of methanol (reagent grade) dropwise, and add 40 mL of n-hexane to dilute the reaction solution. The above suspension was stirred for 30 minutes and then filtered. The filter cake was washed with petroleum ether (50mLx3). The filtrate was collected and concentrated. Compound 3 was obtained by column chromatography (petroleum ether) as an off-white solid (2.57g). Take 32.48g of the compound and add 20 mL of anhydrous tetrahydrofuran to dissolve it under nitrogen protection. Cool in an ice-salt bath and stir for 10 minutes. Measure 3.4 mL of isopropylmagnesium chloride tetrahydrofuran solution (2M) and add it dropwise to the above solution within 5 minutes. Stir and react for 27 minutes. Then add N-methyl-N-methoxy-2-chloroacetamide in toluene dropwise. Solution (0.92g dissolved in 8mL anhydrous toluene). After the dropwise addition, continue the reaction under cooling in the ice-salt bath for 15 minutes, remove the ice-salt bath, and naturally raise the temperature to react for 4 hours. TLC shows that the reaction is completed (PE: EA5/1, RF value 0.3). Add 4N hydrochloric acid dropwise to the reaction solution to adjust the pH to about 1-2, add 20 mL of methyl tert-butyl ether and 20 mL of saturated saline, and shake to separate the liquid. Dry the organic phase over anhydrous magnesium sulfate for 5 minutes, filter, concentrate and evaporate tetrahydrofuran and methyl tert-butyl ether to dryness. Add n-hexane-dichloromethane (20:1, 22mL) to the resulting oil, heat and stir in a 70°C oil bath. Reflux for 30 minutes, then naturally cool to room temperature, filter, and wash the filter cake with cold n-hexane (5 mLx3), and dry under vacuum to obtain compound 4 as a light yellow solid (1.51 g).
1HNMR(75MHz, CDCl3)δ8.19(d,J=1.1Hz,1H),8.13(d,J=8.0Hz,1H),7.82(d,J=1.5Hz,1H),7.68(d ,J=8.4Hz,2H),7.53(d,J=8.1Hz,1H),4.72(s,2H).
Step S3: First obtain compound 7 according to the following synthesis route;
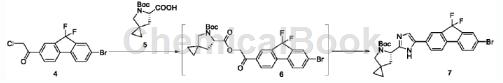
Take 1.44g of compound 4, add 0.98g of proline derivative 5, and 66 mg of potassium iodide, add 20 mL of acetonitrile and stir to disperse evenly at room temperature, add 540 mg of diisopropylethylamine, and stir for 5 hours at room temperature. The dot plate displays The reaction is complete. The reaction solution was evaporated to dryness under reduced pressure, and 20 mL of ethyl acetate was added to dissolve the resulting oil, which was washed successively with 1N hydrochloric acid, 5% sodium hydroxide aqueous solution and saturated brine. The organic phase was dried with anhydrous magnesium sulfate-anhydrous sodium sulfate, filtered, concentrated and evaporated. After drying, the crude compound 6 was obtained as brown oil. Add 25 mL of toluene to dissolve the crude product 6 obtained above, add 6.2 g of ammonium acetate, heat and stir the reaction in a 95°C oil bath under nitrogen protection overnight, remove the oil bath, cool the reaction in an ice bath, add 10 mL of ethyl acetate to dilute, and successively use water ( 20 mL), 5% sodium bicarbonate (20 mL), and saturated saline. Separate the liquids, dry the organic phase with anhydrous sodium sulfate-anhydrous magnesium sulfate, evaporate to dryness under reduced pressure, add 15ml of ethyl acetate, stir and reflux for 30 minutes, add 15ml of n-hexane dropwise while refluxing, then stir and naturally cool to room temperature, filter, filter The cake was washed with ethyl acetate-n-hexane (1:5, 5mLx3), and dried under vacuum overnight to obtain product 7 as a brown powdery solid (1.32g).
1HNMR(75MHz, CDCl3)δ10.61(s,1H),8.01(s,1H),8.01(s,1H),8.01(s,1H),7.89(d,J=7.7Hz,1H) ,7.74(d,J=1.3Hz,1H),7.57(d,J=8.1Hz,1H),7.50(d,J=7.6Hz,1H),7.40(d,J=7.9Hz,1H),7.31 (s,1H),5.11(d,J=6.0Hz,1H),3.51(d,J=10.3Hz,1H),3.13(d,J=10.3Hz,1H),2.82(d,J=12.3Hz ,1H),2.35(t,J=10.5Hz,1H),1.50(s,9H),0.90(s,1H),0.75–0.49(m,3H).
Step S4: First obtain compound 12 according to the following synthesis route;
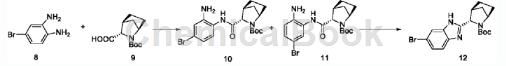
Take compound 8 (768mg) and compound 9 (890mg), add 15mL N,N-dimethylStir and dissolve carboxamide at room temperature, add N-methylmorpholine (830mg) and 2-(7-azobenzotriazole)-N,N,N’,N’-tetramethylurea hexafluorophosphate Ester (HATU, 1.71g), reacted at room temperature. After 1 hour, 150 mL of water was added to the reaction solution, extracted with ethyl acetate (25 mLx3), the organic phase was washed with brine, dried over anhydrous magnesium sulfate and anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to obtain a mixture of 10 and 11 as a reddish-brown oil. Dissolve the above oil in 20 mL of absolute ethanol, seal the tube in an oil bath at 115°C, and heat for 48 hours. The ethanol was evaporated to dryness and purified by column chromatography (petroleum ether-ethyl acetate, 2:1) to obtain compound 12 as a tan oily or foamy solid (1.59g).
1HNMR(75MHz, CDCl3)δ10.72(s,1H),7.59(dd,J=10.9,5.1Hz,1H),7.45–7.19(m,2H),4.54(d,J=2.6Hz, 1H),4.17(s,1H),3.45(d,J=3.4Hz,1H),2.05–1.7(m,6H),1.54
Main reference materials
[1] (CN106117187) Hepatitis C virus inhibitor, pharmaceutical composition and application thereof

 微信扫一扫打赏
微信扫一扫打赏
