
Background and overview[1]
Alipendol, whose chemical name is 2-hydroxy-N-(2-hydroxyethyl)-3-methoxy-5-(2-propenyl) benzamide, is an excellent antispasmodic agent Bile drugs. Pharmacological studies have proven that it has low toxicity, obvious choleretic effect, and can promote the normal production and secretion of bile.
Preparation[1]
At present, there are few public reports on the synthesis of alipendol. Francoi Clemence et al. mentioned a method for preparing alipendol in their patent: using ethyl 2-hydroxy-3-methoxy benzoate as raw material, first react with ethanolamine to obtain 2-hydroxy-3-methoxy -N-(2-hydroxyethyl) benzamide, and then undergo allylation reaction under alkaline conditions of potassium carbonate to prepare 2-(2-propenyloxy)-3-methoxy-N-(2 -Hydroxyethyl) benzamide, and finally alipendol is obtained through Claisen rearrangement reaction. Although this process route is short, the raw material used, ethyl 2-hydroxy-3-methoxybenzoate, is not a common raw material in the market, and its source has not been disclosed. The yield and product purity of each step of the reaction have not been reported. Eunju Kim et al. reported a synthesis method of alipendol: using 2-hydroxy-3-methoxybenzaldehyde (o-vanillin) as raw material, and trimethyl orthoformate under the catalysis of ion exchange resin Amberlyst15 Acetal is generated, and then oxidized and esterified to 2-hydroxy-3-methoxybenzoic acid methyl ester with m-chloroperbenzoic acid and boron trifluoride/diethyl ether; then, through allylation reaction, Claisen Alibendol is prepared through rearrangement reaction and aminolysis reaction. The advantage of this process is that it uses o-vanillin, which is easily available on the market, as raw material, and uses the “one-pot method” to convert aldehydes into esters. The disadvantage is that the reagent used, m-chloroperbenzoic acid, has poor stability, is inconvenient to transport and store, and causes great pollution. , and separation and processing are relatively difficult; all product purification in this process uses column chromatography separation that is difficult to operate in industrial production. Kompella Amala and others used 2-methoxyphenol as raw material, and obtained 2-methoxy-4-(2-propenyl)phenol after allylation reaction and Claisen rearrangement reaction, and then reacted with CO2 React under acidic conditions to obtain 2-hydroxy-3-methoxy-5-(2-propenyl) benzoic acid. This intermediate is then subjected to esterification and aminolysis to obtain the target product-aribenzoic acid. many. This route involves carbonylation, the reaction conditions are harsh, and the yield is low.
Ouyang Hongxia and others used o-vanillin (Ⅱ) as raw material, first obtained 3-methoxy-2-(2-propenyloxy) benzaldehyde (Ⅲ) through allylation reaction, and then used The cheap and easily available inorganic oxidants sodium chlorite and sulfamic acid oxidize the aldehyde into the corresponding acid (IV) in a mixed solvent of acetone-water, and then undergo a methyl esterification reaction to obtain 3-methoxy-2-( 2-propenyloxy) methyl benzoate (Ⅴ), and finally the target product – alibendo (Ⅰ) is obtained through rearrangement and aminolysis reaction. The raw materials of this route are easy to obtain, overcome many shortcomings of the above methods, and can be easily industrialized.
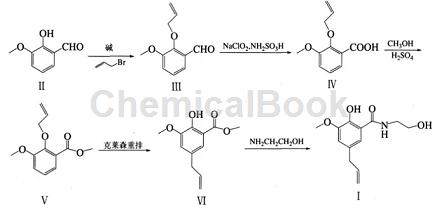
1. Synthesis of 3-methoxy-2-(2-propenyloxy)benzaldehyde (III)
In a 50 mL three-necked flask, add 2. 90 g (19. 1 mmol) o-vanillin (Ⅱ) and 20 mL anhydrous DMF. After complete dissolution, use an ice bath to cool to 0~5°C. Then add 0.53 g (22.9 mmol) NaH to the solution. The solution quickly turns yellow. After stirring for about 10 minutes, add 4.62 g (38.2 mmol) allyl bromide. The reaction is stirred at room temperature for about 3 hours. TLC traces the results. Vanillin until the reaction is complete. After the reaction is completed, add ice water to the reaction solution, then extract the water layer 3 to 4 times with ethyl acetate, combine the organic layers, and concentrate the organic phase under reduced pressure to obtain a yellow oil. Add ethyl acetate ( 2 mL) to dissolve, then filter the solution through a 6 cm thick silica gel layer, rinse completely with ethyl acetate (30 mL), collect the filtrate, and distill the filtrate under reduced pressure to remove the solvent to obtain 3.55 g of yellow liquid (Ⅲ) , the yield is 97%, and the GC purity is 95%. IR (KBr coating method), ν/cm-1: 1691, 1584, 1475, 1382, 1314, 1265, 1062, 982, 905, 785, 760; 1HNMR (600 MHz, CDCl3), δ: 10. 44 (s, 1H), 7. 42 (dd, J = 6.6, 2. 4 Hz, 1H), 7. 15~ 7. 13 (m, 2H), 6. 10~6. 05 (m, 1H), 5. 35 (m, 1H), 5. 37~5. 34 (m, 1H), 4. 67~4. 65(m,2H), 3.90(s,3H).
2. Synthesis of 3-methoxy 2-(2-propenyloxy) methyl benzoate (V)
Add 3.42 g (17. 8 mmol) compound III into a 100 mL three-necked flask, add 30 mL acetone and 15 mL water in sequence, cool to 0~5 ℃ with an ice bath, and add 3. 45 g (35 . 6 mmol) sulfamic acid and 2.53 g (28. 0 mmol) sodium chlorite, continue to react for 1 hour at 0-5°C. After the reaction is completed, use CH2Cl2 Extraction, remove the lower organic layer and wash it thoroughly with saturated brine until neutral, separate the liquids, dry the organic layer with MgSO4 , filter, and evaporate CH2 under reduced pressure Cl2, to obtain white crystal IV, melting point: 55~56 ℃. Put this white crystal into a 100 mL three-necked flask and dissolve it with 10 mL methanol. Then add about 2. 63 g (26. 3 mmol, mass fraction 98%) concentrated sulfuric acid, stir at room temperature overnight, and follow compound IV with TLC until the reaction is complete. After the reaction is completed, add saturated NaHCO3 solution to the reaction solution until it becomes neutral, distill under reduced pressure to remove excess methanol, and extract the residue with anhydrous ether three times. Collect the organic layer, dry it with MgSO4, filter, and reduce Anhydrous ether was removed by pressure distillation, and n-hexane (10 mL) was added to the residue.Heat, stir, and filter while hot. Repeat the operation twice, combine all the filtrate, and concentrate to obtain 3.75 g of colorless liquid (V), with a yield of 95% and a GC purity of 94%. 1HNMR (600 MHz, CDCl3 ), δ: 7. 32 (dd, J = 7. 8, 1. 8 Hz, 1H), 7. 09~ 7. 03 (m, 2H), 6. 15~6. 10 (m, 1H), 5. 38~5. 35 (m, 1H), 5. 23~5. 21 (m, 1H), 4. 57~4. 56 (m, 2H), 3. 88 (s, 1H), 3. 86 (s, 1H).
3. Synthesis of 2-hydroxy-3-methoxy-5-(2-propenyl) methyl benzoate (VI)
Put 2.86 g (12.9mmol) compound V into a 100 mL flask, heat to 220~250°C, react for 2 hours, stop heating, cool to room temperature, add ethyl acetate (15 mL), and add Remove the color with a small amount of carbon powder and filter. The filtrate is washed with saturated brine (5 mL×2). The organic layers are combined and the solvent is distilled off under reduced pressure to obtain a crude product. The crude product is recrystallized with diethyl ether to obtain 2.66 g of white solid (VI). , the yield was 93%, and the GC purity was 97%. Melting point: 44. 6 ~ 45. 9 ℃ (literature value 45 ~ 46 ℃).
4. Synthesis of 2-hydroxy-N-(2-hydroxyethyl)-3-methoxy-5-(2-propenyl)benzamide (Ⅰ)
Add 2. 62 g (11. 8 mmol) VI and 25 mL chloroform into a 50 mL three-necked flask, then add excess ethanolamine, heat to 120°C, and react for 1. 5 h. After the reaction is completed, the reaction is extracted with ethyl acetate. liquid three times, collect the organic phase and wash with dilute hydrochloric acid until neutral, separate the liquids, dry the organic layer with anhydrous MgSO4, filter, and rotary evaporate under reduced pressure to obtain a crude product. The crude product is recrystallized with benzene to obtain a white solid (Ⅰ) 2. 49 g, yield 84%, GC purity 98%. Melting point: 96. 5 ~ 97. 8 ℃ (literature value 95 ~ 96 ℃); 1HNMR (600 MHz, CDCl3) δ: 11. 30 (s, 1H), 7. 09 (br s, 1H), 6. 92 (d, J = 1. 2 Hz, 1H), 6. 81 ( d, J = 1. 2 Hz, 1H), 5. 95~5 . 90 (m, 1H), 5. 09~5. 06 (m, 2H), 3. 89 (s, 3H), 3. 85 (t, J =4. 8 Hz, 2H), 3. 63 ( q, J = 5. 2 Hz, 2H), 3. 32 ( d, J = 6. 6 Hz, 2H), 2. 54 ( brs, 1H). MS (ESI), m /Z: 274 [M + 23].
Main reference materials
[1] Ouyang Hongxia, Zhang Xiaolin, Ding Yonghong, Sun Wuchen. Synthesis of choleretic drug alipendol [J]. Fine Chemicals, 2011, 28(12): 1199-1202.

 微信扫一扫打赏
微信扫一扫打赏
