
Background and overview[1][2]
Rivastigmine bitartrate (IV) is an anti-Alzheimer’s drug developed by the Swiss Novartis Company, also known as Esnen. It is a carbamate-based reversible cholinesterase inhibitor and belongs to the third generation. A drug that improves the function of the choline system. In addition to inhibiting acetylcholinesterase, it can also inhibit butyrylcholinesterase, exerting a dual inhibitory effect. The effect is accurate and stable. It was launched in Switzerland in 1997. In June 2000, my country’s State Food and Drug Administration officially approved rivastigmine (IV) bitartrate to enter the Chinese market.
(S)-3-[1-(Dimethylamino)ethyl]phenol (III) is the key intermediate for the hypoglycemic drug rivastigmine bitartrate. In the prior art, (S)-(+)-camphor-10-sulfonic acid is usually used as a resolving agent. However, in order to obtain products with qualified optical activity, there is a recrystallization phenomenon, and there are problems such as cumbersome operation, high cost, and low recovery rate, which is not conducive to industrial production.
Apply[2-3]
(S)-3-[1-(Dimethylamino)ethyl]phenol (Ⅲ) is the key intermediate for the synthesis of rivastigmine and its tartrate from the antidiabetic drug rivastigmine bitartrate. Intermediate, some studies use m-methoxyacetophenone as the starting material, and then reduce it to obtain 1-(3-methoxyphenyl)ethanol; then chlorine it to obtain 1-(chloroethyl)-3-methoxy Benzene; react with dimethylamine hydrochloride to obtain 1-(3-methoxyphenyl)-N, N-dimethylethylamine; demethylate to obtain 3-[1-(dimethylamino)ethyl]phenol ; Then separate it with (s)-(+)-camphor-10-sulfonic acid to form a salt, recrystallize, and dissociate to obtain (s)-3-[1-(dimethylamino)ethyl]phenol.
Then use ethylamine as raw material, react with ethyl formate to obtain formylethylamine; then react with phosphorus oxychloride to obtain the imine intermediate; reduce sodium borohydride to obtain methylethylamine; then react with triphosgene to obtain N -Methyl-N-ethylformyl chloride. Finally, (s)-3-[1-(dimethylamino)ethyl]phenol is condensed with N-methyl-N-ethylformyl chloride, and then salted with L-tartaric acid to obtain rivastigmine tartrate. This method has easy-to-obtain raw materials, simple operation, low cost, high yield, and little pollution. It is currently a brand-new synthesis route.
There are also studies using (R,S)-3-(1-(dimethylamino)ethyl)phenol as raw material and separation with S-(+)-camphorsulfonic acid to obtain enantiopure ( S)-3-(1-(dimethylamino)ethyl)phenol; then docked with N-ethyl-N-methylcarbamoyl chloride to obtain rivastigmine free base, and finally combined with L-(+)-tartaric acid to form Salt gives rivastigmine bitartrate. By processing the raw material synthesis process, using acid-base alternating impurity removal in the intermediate refining, and using acetone beating for later purification and other process controls, a method for preparing rivastigmine bitartrate was obtained. This method not only improves product purity and yield The efficiency can make the isomer impurities more effectively controlled, and the cost can be greatly reduced, the process can be simplified, and it is more suitable for industrial production.
Preparation [1,4]
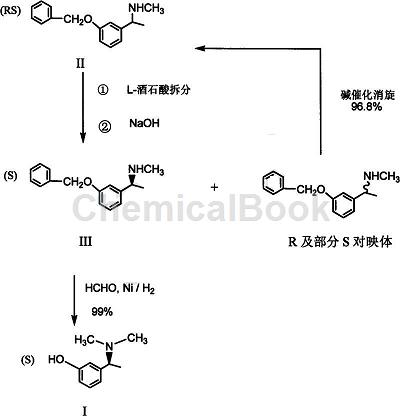
Method 1: The method for preparing (S)-3-(1-dimethylaminoethyl)phenol (I) includes:
1) (RS) m-benzyloxy-α, N-dimethylbenzylamine (II) is salted with L-(+)-tartaric acid in an alcohol solvent or ethyl acetate, crystallized, filtered, and separated to obtain (S)-m-benzyloxy-α, N-dimethylbenzylamine L-tartrate is then subjected to alkali analysis to obtain (S)-m-benzyloxy-α, N-dimethylbenzylamine (III);
2) (S)-m-benzyloxy-α, N-dimethylbenzylamine (III) is added to the alcohol solution with formaldehyde solution or polyformaldehyde, catalyzed by Ray’s nickel, and hydrogenated at room temperature, normal pressure or low pressure. Aminomethylation and simultaneous removal of benzyl group yield (S)-3-(1-dimethylaminoethyl)phenol (I);
3) Step (1) Separate the separation mother liquor after crystallization of (S)-m-benzyloxy-α, N-dimethylbenzylamine L-tartrate. After alkali analysis, free (R) and part of (S) configuration m-benzyloxy-α, N-dimethylbenzylamine base is reacted in an organic solvent with a base as a catalyst at 95-105°C for 1-3 hours to obtain (RS) m-benzyloxy group -α,N-dimethylbenzylamine (II).
Method 2: The separation method of (S)-3-[1-dimethylaminoethyl]phenol (III) includes: dividing a certain proportion of (S)3-[1-dimethylaminoethyl]phenol (III) [Basic] phenol (II) and dinaphthyl phosphoric acid (I) are carried out in an organic solvent under the conditions of 0℃-80℃ and crystallization reaction under normal pressure: after at least 8 hours, the reaction liquid is suction filtered to obtain a filter cake, and then The filter cake is subjected to a salt decomposition reaction in an alkaline solution under the conditions of 0℃-100℃ to obtain (S)-3-[1-dimethylammonium chloride.��Ethyl]phenol (III). The specific operation steps are: add 15kg of absolute ethanol to 6.00kg of (S) 3-[1-dimethylaminoethyl]phenol (II), stir and dissolve at 35°C, and slowly add it dropwise after the reaction solution is completely dissolved. A mixed solution composed of 8.86kg of dinaphthyl phosphoric acid (I) and 9.00kg of absolute ethanol was mechanically stirred in a stirring device.
During the stirring process, solids continued to precipitate, and the solids were (S)-3-[1-dimethylaminoethyl]phenol dinaphthyl phosphate. After reacting for 8 hours, the reaction solution was suction-filtered to obtain 12.00kg of wet product. Dissolve 10kg of low-temperature dried (S)-3-[1-dimethylaminoethyl]phenol dinaphthyl phosphate into 240kg of water, then place it in a stirring device for mechanical stirring, cool to 5°C, and add the concentration dropwise 12L of 0.1% sodium hydroxide solution. During the dripping process of the sodium hydroxide solution, the temperature was kept not higher than 5°C. After the dripping was completed, a large amount of solids precipitated after 12 hours of stirring and the reaction solution was suction-filtered to obtain 4.7kg of wet product. The wet product was placed in a room temperature vacuum drying oven and dried to obtain 2.5 kg of (S)-3-[1-dimethylaminoethyl]phenol (III) finished product. The optical purity of the finished product was 98.76%.
Main reference materials
[1] CN201510728607.6 Resolution method of (S)-3-[1-(dimethylamino)ethyl]phenol (Ⅲ)
[2] CN200910011751.2 Preparation method and application of rivastigmine bitartrate
[3] CN201610896773.1 A preparation method of rivastigmine bitartrate
[4] Preparation method of CN200910055549.X (S)-3-(1-dimethylaminoethyl)phenol

 微信扫一扫打赏
微信扫一扫打赏
